Comité de Ética
El Comité Institucional de Ética en Investigación (CIEI), es una organización integrada por miembros profesionales multidisciplinarios e independientes, quienes protegen los derechos, el respecto a la dignidad, bienestar y seguridad de pacientes que ingresan a un estudio de investigación.
Comité de Ética
El Comité Institucional de Ética en Investigación (CIEI), es una organización integrada por miembros profesionales multidisciplinarios e independientes, quienes protegen los derechos, el respecto a la dignidad, bienestar y seguridad de pacientes que ingresan a un estudio de investigación. Esta tarea implica el análisis riguroso de los ensayos clínicos, la evaluación del balance entre los riesgos y beneficios, así como, la evaluación ética y metodológica de un protocolo de investigación.

El Comité Institucional de Ética evalúa los proyectos basándose en cuatro principios éticos:
- Respeto a la autonomía de los seres humanos
- Beneficencia
- Justicia
- No maleficencia
El CIEI conduce sus evaluaciones guiado bajo los principios de la Declaración de Helsinki, Guía de Buenas Prácticas Clínicas y del Reglamento de Ensayos Clínicos en el Perú.
Registros del Comité Institucional de Ética:
Registro Nacional:
El Comité cuenta con código de registro del INS RCEI-6.
Registro Internacional:
El CIEI está registrado en la OHRP (Office for Human Research Protections) con código institucional IORG0001304 y código del IRB00001733 (Institutional Review Board) ambos con vencimiento 13/set/2025. Además cuenta con el código de garantía federal FWA (Federalwide Assurance) FWA00001219 (vencimiento 31/ene/2029).
Red de Comités de Ética en Investigación (REDCEI)
Nuestros resultados:
Tenemos la experiencia de contar con un equipo autónomo y multidisciplinario que garantiza el buen desempeño en la evaluación de ensayos clínicos y de estudios observacionales como epidemiológicos, siguiendo los principios bioéticos y realizando un balance entre los riesgos y beneficios que tendrá el sujeto de investigación.
En los últimos años hemos evaluado ensayos clínicos de laboratorios farmacéuticos reconocidos como AMGEM inc, Laboratorios Novartis, Sanofi-Aventis, Pfizer, Merck Sharp & Dohme, AstraZeneca, Roche Farma, Bristol Myers Squibb, Johnson & Jonhson, Cerexa Pharmaceutical, Takeda Pharmaceutical, Grunenthal y Bayer. Y sus representantes en el Perú como: ICON Clinical Research, IQVIA RDS Perú, PPD Perú SAC, Paraxel, Inventiv Health, Covance Perú, Resolution Latin America, entre otros.






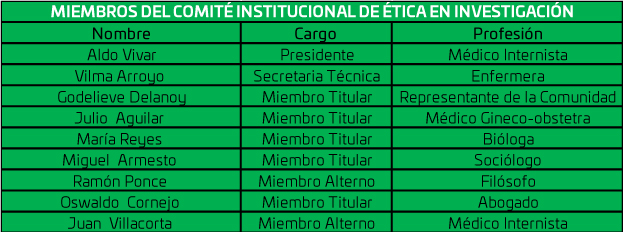
Directorio del Comité de Ética en Investigación
El Directorio está conformado por un equipo multidisciplinario altamente calificado para llevar a cabo una evaluación adecuada a los proyectos de investigación.
DIRECTORIO
Para la ejecución de sus funciones, el Comité Institucional de Ética en Investigación está compuesto por ocho miembros permanentes, solo uno de ellos es miembro suplente, sin tiempo límite de vigencia.
Funciones del Comité
Requisitos para la revisión de un ensayo clínico
Documentos a presentar para revisión inicial:
Protocolo de Investigación:
- Protocolo versión inglés y español
- Justificación para Uso de Placebo (en caso aplicara – versión inglés y español)
Consentimiento(s) Informado(s):
- Consentimiento(s) Informado(s) versión inglés y español.
Manual del Investigador:
- Manual del Investigador versión inglés y español.
Materiales para pacientes:
- Materiales para el participante: encuestas, cuestionarios, material informativo, etc. versión inglés y español.
Documentos del investigador principal (PI) y centro de investigación:
- CV y copia simple del título o carné del Colegio Médico y habilitación vigente del equipo médico del estudio.
- Capacitación en Ética en la Investigación y Capacitación en Conducta Responsable del equipo.
- Declaración de Confidencialidad del Investigador Principal y equipo. (Anexo 9 del MAPRO).
- Declaración de Ausencia de Conflicto de Interés del Investigador principal y equipo de investigación. (Anexo 10 del MAPRO)
- Declaración del Investigador (Anexo 11 del MAPRO).
- Póliza de Seguro vigente
- RD de Autorización de OGITT-INS
- Permiso o autorización de funcionamiento del centro de investigación otorgado por máxima autoridad de la institución al que pertenece.
IMPORTANTE:
Todos los documentos para solicitud de revisión y/o aprobación inicial al CIEI deberán someterse con una carta dirigida al Dr. Aldo Vivar Mendoza, Presidente del Comité de Ética, ser firmada por el Investigador Principal que conduce el estudio de investigación. El documento de sometimiento debe contar con la siguiente información:
- Título y Código del Ensayo Clínico.
- Nombre y Tipo del producto de investigación.
- Objetivo y justificación breve del estudio de investigación.
- Breve cuadro de matriz operacional.
- Tiempo de duración estimado del estudio.
- Número de participantes a enrolar en Perú y número de participantes estimados a enrolar en el Centro de investigación.
- Mecanismo de reclutamiento o enrolamiento de participantes al centro de investigación
- Datos del contacto del Centro de investigación, RCI y Patrocinador
Documentos a presentar para revisión de enmiendas:
- Presentar listado de los nuevos cambios de la enmienda.
- Justificación de los cambios presentados.
- Protocolo y/o Consentimiento Informados con el control de cambios. (En versión español y en idioma original si fuese distinto al español).
- Protocolo y/o Consentimiento Informados con el control de cambios. (En versión español y en idioma original si fuese distinto al español).
- Presentar Oficio de la OGITT-INS en caso de nueva enmienda por las observaciones realizadas por la entidad regulatoria.
* Fecha límite de recepción de documentos hasta 7 días antes de la sesión.
Requisitos para la revisión de un estudio observacional
Documentos a presentar para revisión inicial:
Protocolo de Investigación:
- Protocolo versión español.
Consentimiento Informado y Materiales para pacientes:
- Consentimiento(s) Informado(s) versión en español.
- Materiales para el participante: encuestas, cuestionarios, material informativo, etc. Versión en español.
Documentos del investigador principal (PI) y centro de investigación:
- CV y copia simple del título o carnet del Colegio médico y habilitación vigente del equipo médico del estudio.
- Capacitación en Ética en la Investigación y Capacitación en Conducta Responsable del equipo.
- Declaración de Confidencialidad del Investigador Principal y equipo. (Anexo 9 del MAPRO).
- Declaración de Ausencia de Conflicto de Interés del Investigador principal y equipo de investigación. (Anexo 10 del MAPRO)
- Declaración del Investigador (Anexo 11 del MAPRO).
IMPORTANTE:
Todos los documentos para solicitud de revisión y/o aprobación inicial al CIEI deberán someterse con una carta dirigida al Dr. Aldo Vivar Mendoza, Presidente del Comité de Ética, ser firmada por el Investigador Principal que conduce el estudio de investigación. El documento de sometimiento debe contar con la siguiente información:
- Nombre y Tipo de Estudio de investigación.
- Objetivo y justificación breve del estudio.
- Breve cuadro de matriz operacional.
- Lugar y Tiempo de duración estimado del estudio.
- Número de pacientes estimados a enrolar en el Centro de investigación.
- Mecanismo de reclutamiento o enrolamiento de participantes al estudio.
- Datos del contacto del Centro de investigación.
* Fecha límite de recepción de documentos hasta 7 días antes de la sesión.
Documentos a presentar para revisión de enmiendas:
- Presentar listado de los nuevos cambios de la enmienda.
- Justificación de los cambios presentados.
- Protocolo y/o Consentimiento Informados con el control de cambios. (En versión español y en idioma original si fuese distinto al español).
- Protocolo y/o Consentimiento Informados con el control de cambios. (En versión español y en idioma original si fuese distinto al español).
* Fecha límite de recepción de documentos hasta 7 días antes de la sesión.
Cronograma de sesiones CIEI Prisma 2025

Las sesiones ordinarias del Comité Institucional de Ética en Investigación de Prisma se realizan los días martes de manera virtual con una frecuencia de dos veces al mes.
La fecha límite de entrega de los documentos, es de siete (07) días antes de la sesión programada de acuerdo al calendario de sesiones del CIEI Prisma.
TARIFARIO DE REVISIONES AÑO 2025
COMITE INSTITUCIONAL DE ÉTICA EN INVESTIGACIÓN
| Revisión Inicial de un Ensayo Clínico Revisión de un Ensayo inicial incluye la revisión del Protocolo, Consentimiento Informado, Brochure, Materiales para participantes, Currículum Vitae del equipo de investigación, entrenamientos, y la revisión continúa durante el año incluyendo visitas de supervisión si es necesario. | S/. 2,400 |
| Revisión Inicial de un Ensayo Clínico idéntico previamente evaluado por el CIEI (centro adicional) Incluye la revisión del Protocolo, Consentimiento Informado, Brochure, Materiales para participantes, Curriculum Vitae del equipo de investigación y la revisión continúa durante el año. | S/. 1,250 |
| Revisión Inicial de un Estudio de Investigación Observacional Incluye la revisión al Protocolo, Consentimiento Informado, Curriculum Vitae del equipo de investigación, capacitaciones, instrumentos del estudio, etc. | S/. 1,750 |
| Renovación Anual de Ensayo Clínico y Estudio Observacional Incluye el Informe de Avance del estudio | S/. 1,300 |
| Extensión de Tiempo al Ensayo Clínico
| S/. 1,400 |
| Revisión de Enmienda al Protocolo y al Formato de Consentimiento Informado | S/. 900 |
| Revisión de documentos adicionales, presentados posterior a la revisión inicial, pudiendo ser: Materiales para participantes, Diarios, Cuestionarios, Brochures, entre otros. | S/. 500 |
| Revisión por cambios administrativos o menores al Protocolo y FCI
| S/. 500 |
| Revisión por cambio o inclusión de Investigador Principal y/o Sub-Investigador y cambio de centro de investigación. | S/. 450
|
| Todos los precios incluyen el IGV |
Instructivos, guías y formatos
- Anexo 3: Pautas para elaboración del Consentimiento Informado
- Anexo 4: Informe de Avance / Final para Ensayos Clínicos
- Anexo 5: Informe de Avance / Final para Estudios Observacionales
- Anexo 6: Guía para la Revisión Inicial de la Estudios con Seres Humanos
- Anexo 13: Pautas para elaboración de un Protocolo de Investigación
- Anexo 14: Formato de Supervisiones (FOR-OGITT-066)
CONTACTO
HORARIO DE ATENCIÓN
De lunes a viernes:
- Mañana: 9:00 a.m. a 1:00pm.
- Tarde: 3:00 p.m. a 5:00 pm.
Comité Institucional de Ética en Investigación de Prisma ONG
Av. Guardia Civil 1321
Oficina 1501 – Surquillo – Perú
Mónica Mateo
E-mail: mmateo@prisma.org.pe
Tel. 2090400 Anexo: 246
Cel. 955 346 426
Anita López
E-mail: alopez@prisma.org.pe
Tel. 2090400 Anexo: 258
Cel. 955 368 591